Che cos’è la Sindrome di Lennox-Gastaut

Un’encefalopatia epilettica dello sviluppo rara e grave ad esordio precoce, caratterizzata dalla triade di deficit cognitivo, tipi multipli di crisi e anomalie tipiche dell’elettroencefalografia (EEG). (Orphanet)
La sindrome di Lennox-Gastaut (LGS) è un disturbo dello sviluppo cerebrale che si accompagna a epilessia e che inizia precocemente sin dai primi anni di vita. Più propriamente è un’Encefalopatia Epilettica, cioè una malattia del cervello dovuta all’epilessia. Questo vuol dire che è l’epilessia stessa ad alterare e a disturbare il normale sviluppo del cervello del bambino.
Per questo la LGS viene anche definita Encefalopatia dello sviluppo ed epilessia (DEE – Developmental Encephalopathy and Epilepsy).
L’ Encefalopatia epilettica è una malattia che danneggia il cervello; le crisi epilettiche che non rispondono al trattamento farmacologico interferiscono con lo sviluppo neuro cognitivo del bambino.
NB: nella EE le ‘crisi’ contribuiscono al deficit cognitivo comportamentale.
L’Epilessia è una malattia neurologica caratterizzata dalla presenza di crisi (manifestazioni motorie, involontarie), che si possono ripetere nel tempo.
NB: Un’unica crisi non fa epilessia!
Con crisi epilettiche polimorfe plurigiornaliere.
La malattia inizia principalmente tra i 3 ei 5 anni di età e l’intera triade si sviluppa nel tempo. In più della metà dei pazienti, la compromissione cognitiva è presente all’esordio della malattia e peggiora nel tempo. I problemi comportamentali sono comunemente osservati e possono complicare il trattamento.
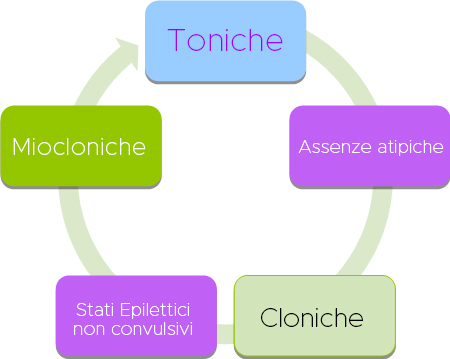
I tipi di crisi caratteristici includono assenze atipiche e crisi toniche durante il sonno, ma comunemente si verificano crisi atoniche durante la veglia, crisi miocloniche, tonico-cloniche e focali, nonché stato epilettico non convulsivo. Le crisi toniche, miocloniche o atoniche possono portare a cadute improvvise (attacchi di caduta).
NB: Le crisi toniche in sonno e le crisi con caduta (toniche o atoniche), sono caratteristiche della sindrome e ne consentono la diagnosi.
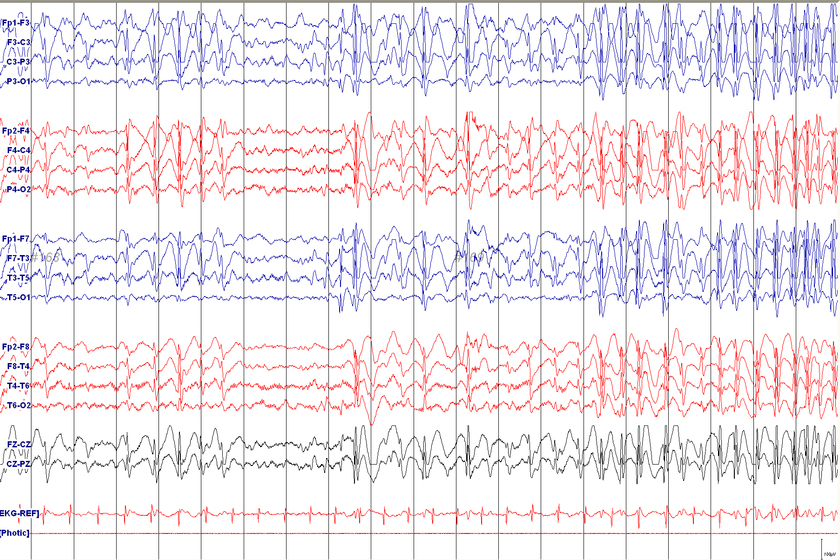
Tramite una valutazione clinica approfondita, un’anamnesi dettagliata del paziente e attraverso tecniche di imaging avanzate, come l’Elettroencefalogramma (EEG) e la Risonanza Magnetica (RMN).
In particolare i segni per diagnosticare la LGS sono:
- Età di esordio delle convulsioni
- Presenza di più tipi di crisi difficili da controllare
- Un Elettroencefalogramma (EEG) che mostra anomalie specifiche.
- Ritardi dello sviluppo e/o disabilità intellettiva (non richieste per la diagnosi ma spesso presenti).
Il trattamento è difficile, la LGS è farmacoresistente e più dell’80% dei pazienti continua a presentare crisi nonostante le terapie.
Le possibilità terapeutiche sono:
- Terapia farmacologica
- Dieta chetogenica (KD)
- Chirurgia: (interventi di resezione, interventi di disconnessione, Interventi di neuro modulazione)
La riabilitazione:
- Logopedia, psicomotricità, terapia occupazionale
- Pet therapy
I farmaci più utilizzati sono:
Viene rivalutata l’efficacia del farmaco somministrato dopo 2-3 mesi
Terapia Chirurgica
(VNS stimolazione del nervo vago, Callosotomia totale o parziale), (DBS stimolazione cerebrale profonda)
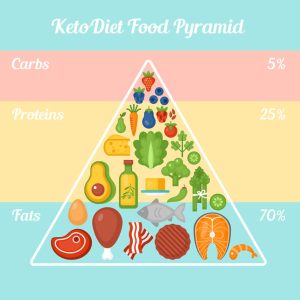
La dieta chetogenica
Un particolare regime alimentare, ricco di grassi e privo di carboidrati e proteine, condotto sotto stretto controllo medico, che ha come obiettivo quello di indurre e mantenere uno stato chetonico
Argomenti correlati










